Vermoeden en doorverwijzing
ASMD herkennen
Voor patiënten met zure-sfingomyelinase deficiëntie (ASMD) kan het tot vijf jaar duren om tot een diagnose te komen¹
Houd in uw differentiaaldiagnose ook zo vroeg mogelijk rekening met ASMD om onzekerheid bij de patiënt weg te nemen en om de schade te beperken.2,3
WEES ALERT OP ASMD ALS U EEN COMBINATIE VAN DEZE VAAK VOORKOMENDE SYMPTOMEN ZIET:

van de patiënten heeft een splenomegalie4

van de patiënten heeft interstitiële longziekte4

van de patiënten heeft een hepatomegalie4

van de patiënten heeft trombocytopenie4
Splenomegalie en hepatomegalie verschijnen vaak als eerste symptomen van ASMD⁴
Als u een van deze vaak voorkomende kenmerken of symptomen ziet, overweeg dan verdere beoordeling op ASMD4
BEOORDELING OP ASMD VOOR DOORVERWIJZING2
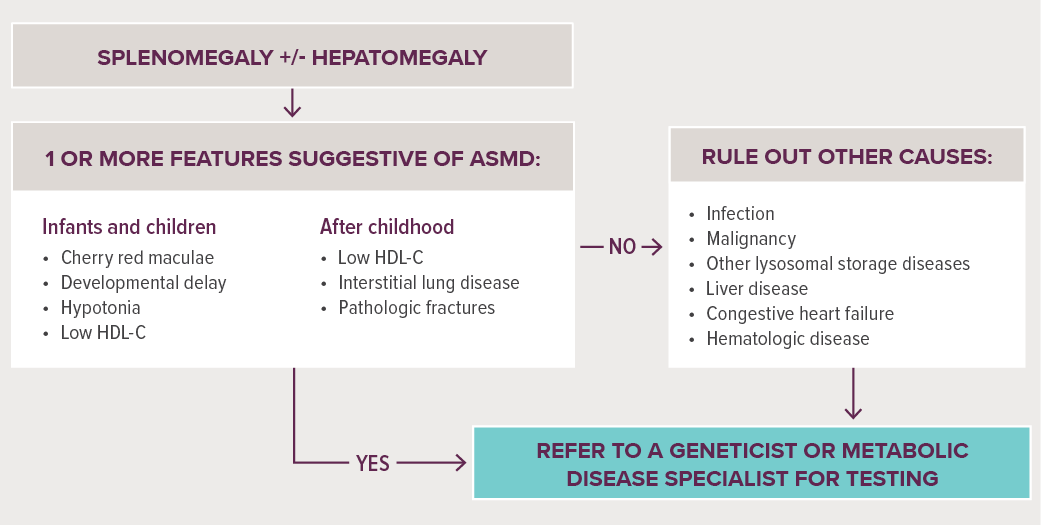
McGovern MM, et al. Genet Med. 2017;19(9)967-974.
Abbreviation: HDL-C, high-density lipoproteïne-cholesterol
Vroege diagnose van ASMD is essentieel voor een goede behandeling en begeleiding ²
Zorg voor doorverwijzing van patiënten met een combinatie van de belangrijkste kenmerken en symptomen
ASMD en de ziekte van Gaucher
Vanwege de overlap van symptomen is het voor een nauwkeurige diagnose raadzaam om parallel te testen op ASMD en de ziekte van Gaucher2
Onder personen met een vermoeden van de ziekte van Gaucher wordt bij elke 4 diagnosen van de ziekte van Gaucher ook 1 patiënt met ASMD gevonden. 5,a
SYMPTOMEN VAN ASMD KUNNEN LIJKEN OP DIE VAN DE ZIEKTE VAN GAUCHER4,6
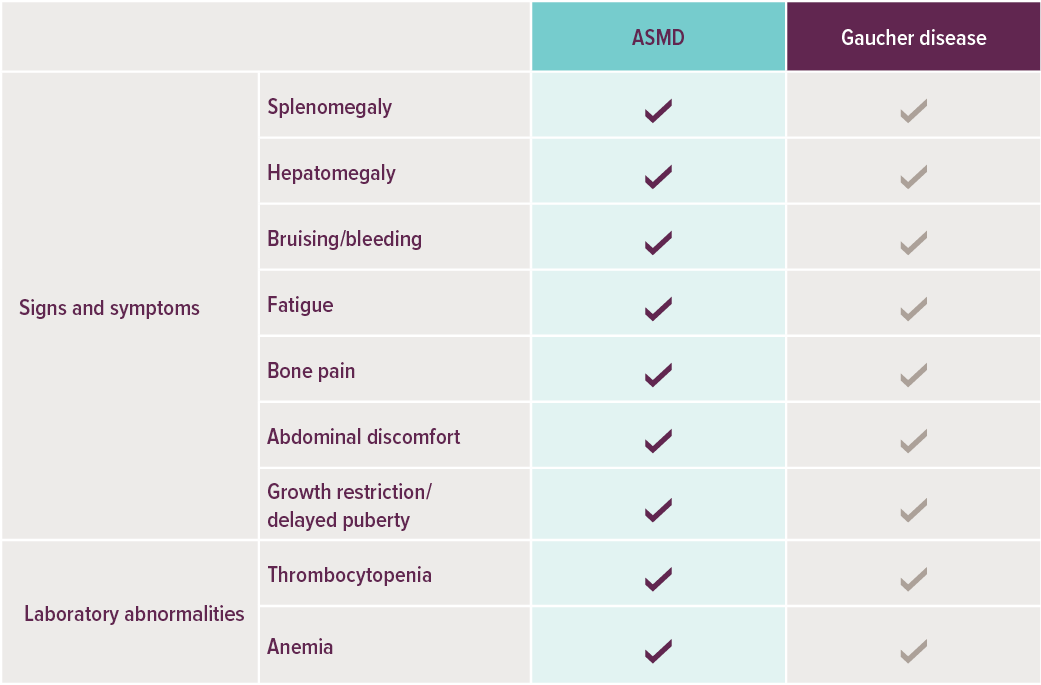
Hoewel ASMD en de ziekte van Gaucher op elkaar lijken, gaat het om een deficiëntie van verschillende lysosomale enzymen2:
- ASMD: deficiënte enzymactiviteit van zure-sfingomyelinase (ASM)2
- Ziekte van Gaucher: deficiënte enzymactiviteit van zure-β-glucosidase2
aOp basis van laboratoriumonderzoeken met DBS samples uit Europa, het Midden-Oosten en Zuid-Afrika.5
Voorkom een late diagnose
Parallel testen op ASMD en de ziekte van Gaucher kan helpen om een late diagnosestelling van patiënten met ASMD te voorkomen2
Referenties: 1. McGovern MM, Wasserstein MP, Giugliani R, et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics. 2008;122(2):e341-e349. doi:10.1542/peds.2007-3016. 2. McGovern MM, Dionisi-Vici C, Giugliani R, et al. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med. 2017;19(9)967-974. doi:10.1038/gim.2017.7. 3. Henderson SL, Packman W, Packman S. Psychosocial aspects of patients with Niemann-Pick disease, type B. Am J Med Genet A. 2009;149A(11):2430-2436. doi:10.1002/ajmg.a.33077. 4. McGovern MM, Avetisyan R, Sanson B-J, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. 2017;12:41. doi:10.1186/s13023-017-0572-x. 5. Lukacs B, Nieves Cobos P, Murko S, Santer R, Kasper D, Wessels C. Multiplexed testing for Gaucher, Niemann Pick A/B disease and acid lipase deficiency. Poster presented at: 14th Annual WORLDSymposium; February 5-9, 2018; San Diego, CA. 6. Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis and assessment: consensus statements. Eur J Pediatr. 2004;163(2):58-66. doi:10.1007/s00431-003-1362-0.